3.8. Relabeling Existing Data within the Database
In this tutorial, we will pursue calculations in the vein of the previous labeling example, but we will now show how to relabel data that is already present in the database, and how to add the newly labeled data back into the database. We will also show how to use the subdivided relabeling approach to run large dataseets in parallel.
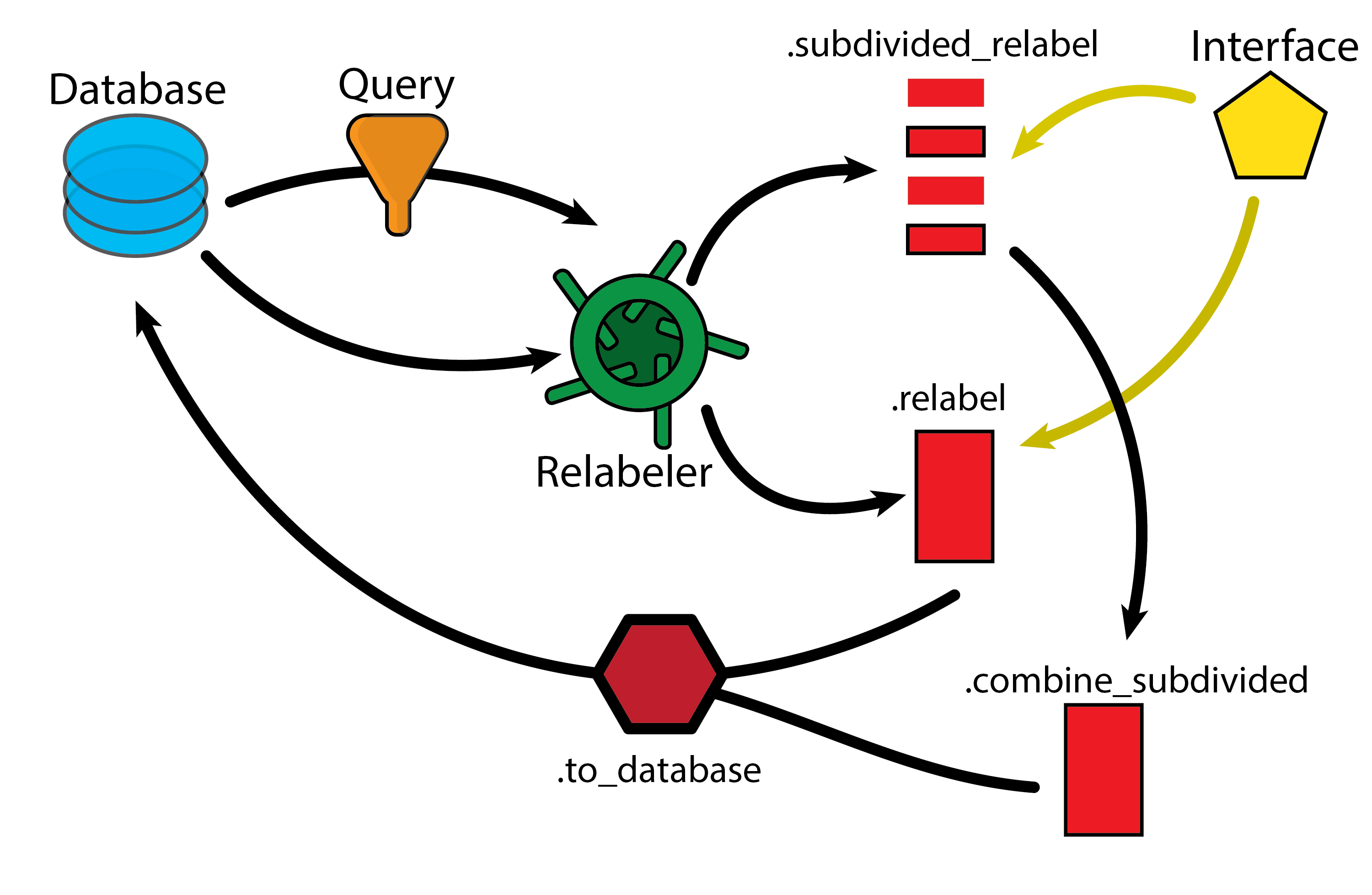
3.8.1. Learning Objectives
Learn how to use the pharmaforge package with ase to re-label data in the database.
Learn how to add the new_data to the database.
Learn how to use the parallel interface to subdivide relabeling tasks into chunks.
3.8.2. Required Files
The tutorial python script is located in examples/Relabeling
3.8.3. Tutorial
As with most of our examples, we first need to import the required packages, and set up a working database. For this exmaple, we will create a temporary database to work with, called “example_add_data” which will have the collection “example_qdpi” within it. This example is actually just working with the same t8 dataset that we used in the previous example, but you can use any dataset that you want.
from pymongo import MongoClient
from pharmaforge.queries.query import Query
from pharmaforge.labeling.relabeler import Relabeler
from pharmaforge.interfaces.xtbio import XTBInterface
from pharmaforge.interfaces.deepmdio import DeepMDInterface
from pharmaforge.recipes.GeneralDatabase import GeneralRecipe
# The first part of this example just uses the hdf5 file to create an example database. This is identical to the example in the setup_smiles.py file, and is the same as the t8 database.
# This is just to have a different working database to work with and test.
try:
client = MongoClient('mongodb://localhost:27017/')
except:
print("MongoDB is not running, or you haven't created the database in the previous example. Please start MongoDB and try again.")
exit()
# Generate a collection with some data
if "example_add_data" in client.list_database_names():
print("Database already exists, dropping it.")
client.drop_database("example_add_data")
# Create a new database from the hdf5 file
print("Creating a new database from the hdf5 file.")
# Now we can read the new file and create a collection.
GeneralRecipe("example_add_data",
"mongodb://localhost:27017/",
input_dir="inputs/",
spin=0,
level_of_theory= "wB97XM-D3(BJ)/def2-TZVPPD",
basis_set= "def2-TZVPPD",
functional= "wB97XM-D3(BJ)",
data_source= "example_add_data")
# Load the newly created datbase
db = client["example_add_data"]
# END Database Generation
Now, as a start, we will show how to relabel an entire database (with all entries). This is done by using the Relabeler class. In the next code snippet, we initialize the class, add the database to the class as the data to be relabeled, and then initialize the interface (in this case XTB) which we will use to relabel the data.
# Call the relabeling class
relabeling_task = Relabeler()
# Add data to the relabeling task.
relabeling_task.select_data(db)
# Select the interface to use for the relabeling task.
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
Now, at this point we haven’t done any calculations. We will now set up the calculation and run it, and then add the data back into the database.
# Do the actual relabeling. (This technically returns the data, but we don't need it here.)
# Note that this can take a bit - depending on level of theory and number of workers.
# The arguments after restart are the same as the ones usually used for the calculate method of the interface.
data = relabeling_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
# No data is added to the datbase yet, so we need to do that. This matches by the formula (molecule_id) and adds the data to the database.
relabeling_task.to_database(db)
Note - that now the data is present within the database, and you can query it as you would any other data.
3.8.3.1. Relabeling only Query
As is described elsewhere, you can also relabel only a query. This is done in a similar way to the above, but now you pass a query to the class alongside the datbase.
# Now, we can try this with a query, which will select only two calculations to do.
q = Query("molecule_id eq C10H10O0N10")
results = q.apply(db, 'example_qdpi')
print("real_qdpi", results[0].get("theorylevels")['wB97XM-D3(BJ)/def2-TZVPPD']['energies'])
new_task = Relabeler()
new_task.select_data(db, query="molecule_id eq C10H10O0N10")
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
data = new_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
print("xtb", data['example_qdpi']['energy'])
new_task = Relabeler()
new_task.select_data(db, query="molecule_id eq C10H10O0N10")
interface = DeepMDInterface(model="qdpi.pb")
data = new_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
print('deep', data["example_qdpi"]['energy'])
print("Relabeling task completed, and two calculations were done based on the query.")
exit()
Note - here we have not added the data back into the datbase, but you COULD do this if you wanted to.
3.8.3.2. Chunking Large Relabeling Tasks
Now, often you will have a large dataset and maybe a more expensive interface than DFTB, this can be used with the Relabeler class to chunk the relabeling task into smaller pieces that can either be run locally (as in this example), or moved to a cluster or other parallel high-throughput computing system.
# Now for BIG datasets, we can use the subdivided relabeling task.
par_task = Relabeler()
par_task.select_data(db)
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
par_task.subdivided_relabel(nchunks=2)
# This step could be done elsewhere, but we will do it here for the sake of the example.
# Note - this does NOT require anything other than the chunk files, so you can do this on a different machine if you want.
Relabeler.relabel_chunk("chunk_0.pckl", interface=interface,limit=0,verbose=False,num_workers=1)
Relabeler.relabel_chunk("chunk_1.pckl", interface=interface,limit=0,verbose=False,num_workers=1)
# Note - the same calculations could be launched using the run-chunk-relabel script that ships with the package. For that, you have to provide the interface arguments
# in the form of a key value pair file.
# For example like this:
# <test.args file>
# method=GFN2-xTB
# accuracy=1.0
# electronic_temperature=300.0
#<end file>
# Then you can run the script like this:
# run-chunk-relabel -p chunk_0.pckl -i XTB -k test.args -n 1
# This will run the relabeling task for the first chunk in parallel using 1 worker.
# Importantly, this DOES NOT require the database to be running - just the package has to be installed with whatever interface you
# want to use.
combine_task = Relabeler()
data = combine_task.combine_subdivided(["chunk_0_relabel_GFN2-xTB.pckl", "chunk_1_relabel_GFN2-xTB.pckl"])
Note, the general logic here is to run the setup for a parallel task on a local system, generate pickle (binary) files that describe the tasks that can be run on a parallel system (which must also have pharmaforge installed, but need not have the database present), and then run the tasks there which then generate output pickle files. These output pickel files can then be read back into the local system and combined using the pharmaforge.relabeling.Relabeler.combine_subdivided function.
As with before, after this point you can add the data back into the database if you want using the same to_database method as before.
3.8.4. Full Code
from pymongo import MongoClient
from pharmaforge.queries.query import Query
from pharmaforge.labeling.relabeler import Relabeler
from pharmaforge.interfaces.xtbio import XTBInterface
from pharmaforge.interfaces.deepmdio import DeepMDInterface
from pharmaforge.recipes.GeneralDatabase import GeneralRecipe
# The first part of this example just uses the hdf5 file to create an example database. This is identical to the example in the setup_smiles.py file, and is the same as the t8 database.
# This is just to have a different working database to work with and test.
try:
client = MongoClient('mongodb://localhost:27017/')
except:
print("MongoDB is not running, or you haven't created the database in the previous example. Please start MongoDB and try again.")
exit()
# Generate a collection with some data
if "example_add_data" in client.list_database_names():
print("Database already exists, dropping it.")
client.drop_database("example_add_data")
# Create a new database from the hdf5 file
print("Creating a new database from the hdf5 file.")
# Now we can read the new file and create a collection.
GeneralRecipe("example_add_data",
"mongodb://localhost:27017/",
input_dir="inputs/",
spin=0,
level_of_theory= "wB97XM-D3(BJ)/def2-TZVPPD",
basis_set= "def2-TZVPPD",
functional= "wB97XM-D3(BJ)",
data_source= "example_add_data")
# Load the newly created datbase
db = client["example_add_data"]
# END Database Generation
print("***************************************************")
# Call the relabeling class
relabeling_task = Relabeler()
# Add data to the relabeling task.
relabeling_task.select_data(db)
# Select the interface to use for the relabeling task.
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
# Do the actual relabeling. (This technically returns the data, but we don't need it here.)
# Note that this can take a bit - depending on level of theory and number of workers.
# The arguments after restart are the same as the ones usually used for the calculate method of the interface.
data = relabeling_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
# No data is added to the datbase yet, so we need to do that. This matches by the formula (molecule_id) and adds the data to the database.
relabeling_task.to_database(db)
print("Relabeling task completed and data added to the database.")
print("***************************************************")
# Now, we can try this with a query, which will select only two calculations to do.
q = Query("molecule_id eq C10H10O0N10")
results = q.apply(db, 'example_qdpi')
print("real_qdpi", results[0].get("theorylevels")['wB97XM-D3(BJ)/def2-TZVPPD']['energies'])
new_task = Relabeler()
new_task.select_data(db, query="molecule_id eq C10H10O0N10")
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
data = new_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
print("xtb", data['example_qdpi']['energy'])
new_task = Relabeler()
new_task.select_data(db, query="molecule_id eq C10H10O0N10")
interface = DeepMDInterface(model="qdpi.pb")
data = new_task.relabel(interface=interface,
restart=False,
limit=0,
verbose=False,
num_workers=1)
print('deep', data["example_qdpi"]['energy'])
print("Relabeling task completed, and two calculations were done based on the query.")
exit()
# End query example
print("***************************************************")
# Now for BIG datasets, we can use the subdivided relabeling task.
par_task = Relabeler()
par_task.select_data(db)
interface = XTBInterface(default=True, method="GFN2-xTB",
accuracy=1.0,
electronic_temperature=300.0)
par_task.subdivided_relabel(nchunks=2)
# This step could be done elsewhere, but we will do it here for the sake of the example.
# Note - this does NOT require anything other than the chunk files, so you can do this on a different machine if you want.
Relabeler.relabel_chunk("chunk_0.pckl", interface=interface,limit=0,verbose=False,num_workers=1)
Relabeler.relabel_chunk("chunk_1.pckl", interface=interface,limit=0,verbose=False,num_workers=1)
# Note - the same calculations could be launched using the run-chunk-relabel script that ships with the package. For that, you have to provide the interface arguments
# in the form of a key value pair file.
# For example like this:
# <test.args file>
# method=GFN2-xTB
# accuracy=1.0
# electronic_temperature=300.0
#<end file>
# Then you can run the script like this:
# run-chunk-relabel -p chunk_0.pckl -i XTB -k test.args -n 1
# This will run the relabeling task for the first chunk in parallel using 1 worker.
# Importantly, this DOES NOT require the database to be running - just the package has to be installed with whatever interface you
# want to use.
combine_task = Relabeler()
data = combine_task.combine_subdivided(["chunk_0_relabel_GFN2-xTB.pckl", "chunk_1_relabel_GFN2-xTB.pckl"])
combine_task.to_database(db, level_of_theory="test")
from pharmaforge.queries.query import Query
q = Query("nmols gt 0")
results = q.apply(db, 'example_qdpi')
print(results[0].get("theorylevels").keys())
print("Relabeling task completed, and data added to the database.")
print("***************************************************")
# Clean up the database at the end of the example.
client.drop_database("example_add_data")